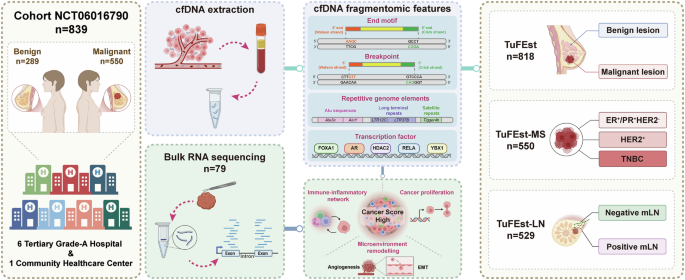
Summarize this content to 100 words: Ethical statementsAll procedures involving human participants complied with all relevant ethical regulations and were conducted in accordance with the Declaration of Helsinki. All datasets were collected under institutional review board approval (Approval No.2023-1048 by the Second Affiliated Hospital of Zhejiang University School of Medicine, Approval No.IIT-20230612-0118-01 by Hangzhou First People’s Hospital, Approval No.2024-KLS-013-02 by Zhejiang Provincial Hospital of Traditional Chinese Medicine, Approval No.2024-033 by Ningbo Medical Center Lihuili Hospital, Approval No.IRB-20230357-R by Women’s Hospital School of Medicine, Zhejiang University, Approval No.2023-0548 by Sir Run Run Shaw Hospital, Approval No.2024-002 by XiangFu Community Health Service Center). All data were de-identified before model development. Written informed consent was obtained from all participants prior to study participation.Study populationThis study was designed as a prospective multicenter case-control study (NCT06016790) that continuously enrolled 839 participants, including 550 BC patients and 289 benign controls from seven Chinese medical institutions: the Second Affiliated Hospital of Zhejiang University School of Medicine (SAHZU), Hangzhou First People’s Hospital (HZFH), Zhejiang Provincial Hospital of Traditional Chinese Medicine (ZJTCM), Ningbo Medical Center Lihuili Hospital (LHLI), Women’s Hospital School of Medicine Zhejiang University (WHZJU), Sir Run Run Shaw Hospital (SRRSH), and Xiangfu Community Health Service Center (XFCHC). Eligible participants were females aged 18-70 years with pathologically confirmed diagnoses of either: (1) primary invasive breast carcinoma or carcinoma in situ, or (2) benign breast lesions (fibroadenoma/adenosis/fibrocystic hyperplasia). All participants provided their plasma samples. Exclusion criteria included pregnancy/lactation, prior malignancies (5-year window), suspected but unconfirmed malignancies within the preceding year, recent blood transfusions (30-day window), active mastitis, and other investigator-determined contraindications.Sample collection and processingPeripheral blood (8–10 mL) was collected in specialized cfDNA tubes (Ardent BioMed Cat. # BY10240301) during routine blood tests or before surgery. Samples were subjected to dual centrifugation (1,600 × g → 16,000 × g, 10 min each at 4 °C) for plasma isolation, followed by immediate storage at −80 °C. Frozen plasma was batch-shipped on dry ice to the central laboratory (OmixScience Research Institute, Hangzhou, China) for processing.cfDNA assays and whole-genome sequencingcfDNA was extracted from a median plasma volume of 1 mL using the VAMNE MagUltra circulating cell-free DNA isolation kit (Vazyme Biotech, Nanjing, China, Cat. # N913) according to the manufacturer’s protocol, followed by precise quantification using a Qubit 4.0 fluorometer (Thermo Fisher Scientific, Lenexa, KS, USA) and storage at −80 °C. For library preparation, 5–20 ng of the extracted cfDNA was processed with the VAHTS Universal DNA library prep kit for Illumina V3 (Vazyme Biotech, Nanjing, China, Cat. # N610) and barcoded using VAHTS multiplex oligos set 5 for Illumina (Vazyme Biotech, Nanjing, China, Cat. # N322). The quality and integrity of the libraries were evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) to confirm that the libraries met the necessary standards for optimal fragment size distribution. The final libraries were sequenced on the NovaSeq X Plus platform (Illumina, San Diego, CA, USA) using paired-end 150 bp reads, with an average coverage depth of 2× for the cfDNA samples.Raw WGS data underwent rigorous quality control beginning with FastQC (version 0.12.1, https://www.bioinformatics.babraham.ac.uk/projects/fastqc) evaluation, followed by quality filtering and adapter trimming using Cutadapt (version 4.5, https://github.com/marcelm/cutadapt) and Ktrim (version 1.4.1, https://github.com/hellosunking/Ktrim). The processed reads were aligned to the human reference genome (GRCh37) using BWA-MEM (version 0.7.17, https://github.com/lh3/bwa) with default settings, ensuring that only high-quality reads with a Phred score above the threshold were retained. Further analyses were restricted to uniquely mapped reads that were free of PCR duplicates. These mapped reads were subsequently sorted and indexed using Samtools (version 1.9, https://github.com/samtools/samtools), and duplicates were identified and eliminated using Picard (version 2.18.29, https://github.com/broadinstitute/picard). The distribution of fragment sizes and relevant genomic features was inferred based on the coordinates of mapped read pairs, following established protocols using Picard. To identify large-scale epigenetic variations in cfDNA fragmentation across the genome, which can be detected with low-coverage WGS, the hg19 reference genome was segmented into non-overlapping 5 Mb bins.Fragmentomic feature identificationWe systematically quantified the frequencies of 4-bp end motifs and 6-bp breakpoint motifs, normalizing total motif occurrences to unity within each category to enable comparative analyses. For the 6 bp motifs, we characterized the 3 bp sequences flanking cfDNA 5’ breakpoints (both upstream and downstream) and established genome-wide distribution profiles. To maintain consistency, the overall frequency of all 6 bp breakpoint motifs was normalized to one. We further organized the motif features into an I × J matrix, where each row corresponds to a participant identified by their sample ID. This matrix format allows for efficient interpretation and comparison of motif patterns within the cohort, leading to deeper insights into cfDNA motif distributions.Regional fragment size analyses employed 100 kb genomic bins using Deeptools (version 3.1.2, https://github.com/deeptools/deepTools), optimized to balance resolution with sufficient read depth (~25,000 reads/bin). Each bin contained short (S, 100–150 bp) and long (L, 151-220 bp) fragments. To ensure data accuracy, regions overlapping the ENCODE blacklist or the hg19 gap track from the UCSC Genome Browser were excluded, as these regions typically show poor mappability. This approach yielded robust S/L ratios for the fragmentation pattern analyses. By focusing on fewer, higher-quality bins, we improved the reliability of our fragment-size distribution analyses.Alu repeats analyses of cfDNAWe categorized all elements from the gene and pseudogene families as transfer RNA (tRNA), signal recognition particle RNA (srpRNA), small nuclear RNA (snRNA), small cytoplasmic RNA (scRNA), and ribosomal RNA (rRNA). Likewise, we grouped elements from the DNA, retroposon, and Rolling Circle (RC) families into the transposable element category. This led to eight primary groups for analysis: LINE, SINE, Medium Repeat (MER), DNA_TcMar-Tigger (DNA Transposable elements), Long Terminal Repeat (LTR), satellites, transposable elements, and RNA elements. In our cfDNA feature analyses and validation, we focused on critical elements, including Alu sequences, long terminal repeats (LTRs), RNA elements, and transposable elements. Through this systematic classification, we adopted a thorough approach to understand cfDNA features, which are essential for investigating their roles in BC. The TuFEst algorithm integrates these repeat elements with the N-index and E-index and incorporates TF features to predict non-cancerous and cancerous status.Machine learning and deep learning algorithm modelIn this study, TuFEst was implemented as a binary classification model50. It outputs a “cancer score” between 0 and 1, representing the probability that a plasma cfDNA sample originates from an individual with cancer. A cutoff of 0.5 was applied to this score to classify samples as “cancer” or “non-cancer.” Although the score is a surrogate measure of the relative burden of tumor-derived DNA and the name suggests its long-term potential for quantification, the current application and validation focus on classification performance.A GLM with a binomial distribution and a logit link function, equivalent to logistic regression, was developed for binary classification. The model utilized fragmentomics features as predictors, with the outcome variable coded as 1 for malignant and 0 for benign status. To address potential sample dependencies, group information was incorporated using SampleID. The analytical pipeline included feature standardization (StandardScaler) followed by logistic regression with a maximum of 1000 iterations. Model performance was assessed through 10-fold stratified cross-validation with shuffling (random state = 42) to ensure reproducibility while maintaining class balance across folds.A SVM with a linear kernel was implemented for classification. The model employed standardized fragmentomics features, with malignant status encoded as 1 and benign as 0. Sample identifiers were retained for group-level analysis. The pipeline consisted of feature scaling followed by SVM classification with probability estimation enabled. Performance was evaluated using 10-fold stratified cross-validation, with metrics including accuracy, AUC, sensitivity, specificity, PPV, and NPV. Probability thresholds at specificity levels of 0.9 and 0.95 were derived from ROC analysis to support clinical decision-making. All predictions and probabilities were systematically recorded for aggregated analysis.An XGBoost classifier was implemented using a reproducible pipeline (random state = 42). Fragmentomics features were standardized prior to model training, with binary outcomes indicating malignant (1) or benign (0) status. The model was evaluated using 10-fold stratified cross-validation, with comprehensive metrics recorded for each fold. Clinical decision thresholds were established by identifying probability values corresponding to 0.9 and 0.95 specificity levels from ROC curves. All predictions and sample metadata were stored for subsequent analysis, and the final pipeline was saved for potential future use.A MLP neural network was developed with fixed random state (42) for reproducibility. Features were standardized before being input to the MLP classifier. The model was evaluated using 10-fold stratified cross-validation, with performance metrics including accuracy, AUC, sensitivity, specificity, PPV, and NPV. Probability thresholds for specific specificity levels (0.9 and 0.95) were determined from ROC analysis to define clinical decision boundaries. The trained pipeline, including preprocessing and classification steps, was preserved for potential application, and overall discriminative ability was validated through aggregated ROC analysis.A GBM was implemented using scikit-learn’s GradientBoostingClassifier. The model was trained on standardized fragmentomics features and evaluated via 10-fold stratified cross-validation (random state = 42). Each fold yielded probability estimates and performance metrics, including accuracy, AUC, sensitivity, specificity, PPV, and NPV. Clinical thresholds were identified by setting specificity levels at 0.9 and 0.95 in ROC analysis. Predictions, true labels, and group identifiers were stored for combined analysis, and the final model was saved for potential deployment.A SEM integrated predictions from five base learners-SVM, GLM, GBM, XGBoost, and MLP-using a logistic regression meta-learner. The pipeline included feature standardization and was evaluated through 10-fold stratified cross-validation. Performance metrics were calculated for each fold, and probability thresholds were established at specificity levels of 0.9 and 0.95. The ensemble demonstrated strong discriminative ability (AUC = 0.943), with all predictions and sample information systematically recorded. The complete pipeline was saved to support potential clinical implementation.For tumor detection and classification, we screened six distinct machine learning algorithms to build the models: GLM, SVM, XGBoost, MLP, GBM, and SEM. These models were rigorously trained and evaluated using bootstrapping combined with 10-fold cross validation. During the 10-fold cross-validation process, the training set was randomly divided into 10 subsets, with nine subsets used for training and the remaining one used for testing, cycling 10 times to ensure that each subset was used for testing once. We only used training cohort samples from benign individuals and cancer patients, training classifiers in the machine learning algorithms with features such as motif feature frequencies, and generating models to predict cancer scores for each sample. Notably, all validation datasets remained untouched during model training. The cancer score ranged from 0 to 1, with a higher score indicating a greater probability of cancer. After the evaluation, we selected the best-performing model for the downstream analyses.These models were subsequently applied to the validation datasets to generate cancer prediction scores for each validation sample and assess the model performance. We compared the AUC values of the different models in the validation cohort and the sensitivity/specificity at a fixed specificity threshold in the internal validation cohort.Three specialized models have been developed for different clinical applications. The TuFEst model was designed to distinguish between benign individuals and cancer patients using a stacked ensemble that incorporates feature information, including TF binding site coverage, to enhance predictive accuracy. The TuFEst-MS model employs a GLM method to classify three BC subtypes: ER+/PR+, HER2+, and TNBC. This classification capability is particularly important for predicting the subtype of metastatic lesions in patients with BC. The TuFEst-LN model utilized GLM to predict whether a patient had mLN, specifically for analyzing patients with radiological evidence of mLN but no surgical confirmation, or those with discordant imaging-pathological results. All models underwent rigorous training using 10-fold cross validation, with prediction scores calculated for each sample. The models were assessed using the AUC-ROC metric to ensure their accuracy and reliability in practical applications.Selection and analyses of differential featuresWe utilized the wilcox.test() function to perform the Wilcoxon rank-sum test, assessing the statistical significance of differences in various features across different classifications. The parameter exact = FALSE indicates the use of an approximate computational method. Features with a p-value less than 0.05 were selected to ensure statistical significance and eliminate the influence of random factors. Additionally, we applied a fold change threshold greater than 1, retaining only features that exhibited significant differences between the two groups. Subsequently, the top and bottom 30 features were identified, and the median ratio between the benign and malignant groups was calculated for each feature. Finally, a standardized feature heatmap was generated using the pheatmap() function from the pheatmap package, while the scale() function was applied for Z-score normalization, enhancing visualization and facilitating further analyses.Genome-wide TF analysesChIP-seq peaks from 5620 experiments were downloaded from the ReMap 2020 database (https://remap2020.univ-amu.fr/download_page), selecting the CRMs (Cis-Regulatory Modules) data type, which includes a total of 1,732,560 peaks.For each autosomal peak, the peak center was designated as position 0, and the mean coverage was computed for each sample within a ± 1,000 bp window relative to the peak center. The processed data were then incorporated as a feature into the model training pipeline to distinguish between benign individuals and patients with cancer.A series of filters were applied to ensure sufficient sequencing depth across benign and cancer samples, retaining only peaks with read counts >25, read coverage ≥0.9, and fold change ≤0.5 or ≥2.To investigate the biological processes associated with the identified TFs, we performed KEGG pathway enrichment analyses using the clusterProfiler package in R. TFs were first mapped to Entrez IDs, and the KEGG enrichment analysis was applied to identify significantly enriched pathways. Pathways were ranked based on their adjusted p-values, and the enriched pathways were visualized in a bubble chart using ggplot2, with bubble size representing gene count and color indicating p-value significance.Estimation of the tumor immune microenvironmentTo comprehensively profile the tumor immune microenvironment, we employed two computational deconvolution methods: CIBERSORT51 and xCell52, which were used to quantify the relative proportions of distinct tumor-infiltrating immune cell populations from the bulk transcriptomic data. In addition, the ESTIMATE algorithm53 was applied to calculate immune and stromal scores, reflecting the overall infiltration levels of immune and stromal components within the tumor tissue.Gene enrichment and pathway analysesGene Set Enrichment Analysis (GSEA) was conducted using the “clusterProfiler” R package54 to identify significantly enriched hallmark gene sets. GSVA was performed using the “GSVA” R package55 to compute sample-wise enrichment scores for specific gene sets. Differentially expressed genes between the Cancer Score-High and Cancer Score-Low groups were identified using the “limma” R package, with thresholds set at |log₂(fold change)| >1 and P < 0.05.Statistics & reproducibilityFor statistical analysis, a ROC curve was generated using the pROC package (v. 1.18.5). Based on the true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) for cancer prediction, we calculated the sensitivity [TP/(TP + FN)], specificity [TN/(Tn + FP)], positive predictive value (PPV) [TP/(TP + FP)], negative predictive value (NPV) [TN/(Tn + FN)], and accuracy [(TP + TN)/(TP + FP + Tn + FN)]. These metrics were computed using the confusion matrix function of the caret package (v. 6.0.94) in R (v. 4.4.1). The preProcess function (v.1.2.2) from the caret package was used for feature standardization. Additionally, the confusion matrix algorithm was employed to compare ROC curves, providing a comprehensive evaluation of the diagnostic test performance and ensuring accurate differentiation between benign and malignant cases. No statistical method was used to predetermine sample size. No data were excluded from the analyses. Sample processing for cfDNA library preparation and sequencing was randomized by case/control status to minimize batch effects. The Investigators were not blinded to allocation during the experiments or during outcome assessment.Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Source link
Fragmentomic liquid biopsy enables early breast cancer detection, molecular subtyping and lymph node assessment
Leave a Comment